CD300A represents a potentially first-in-class therapeutic platform targeting damage-associated molecular pattern (DAMP)-driven inflammatory amplification, a common pathological mechanism shared by multiple acute injuries associated with massive cell death. TNAX103, a humanized anti-CD300A mAb, is an innate immune checkpoint modulator that prevents DAMPs from developing into inflammation. Rather than suppressing immunity, TNAX103 restores the body’s natural ability to rapidly clear dead cells before they trigger harmful inflammation.
Phosphatidylserine (PS) is a phospholipid normally confined to the inner leaflet of the plasma membrane in viable cells. When cells undergo stress or cell death, PS becomes exposed on the outer surface of the plasma membrane. Although cells can die through several distinct mechanisms, one feature is shared among them all: exposure of PS. PS serves as an “eat me” signal, instructing phagocytes to engulf dying cells while minimizing inflammatory responses. If dead or dying cells are not promptly cleared, they eventually undergo secondary necrosis, releasing damage-associated molecular patterns (DAMPs). These DAMPs activate pattern recognition receptors (PRRs) on innate immune cells, triggering sterile inflammation.
Because PS exposure alone does not distinguish between physiological cell turnover and tissue injury, the immune system integrates PS-derived signals with DAMPs-derived signals. CD300a and CD300b function as paired receptors that interpret these opposing cues. Rather than competing for PS binding itself, they integrate signals from both pathways to continuously regulate the balance between inflammation and tissue repair.
CD300a and CD300b are paired receptors.
- Expressed primarily on myeloid cells
- Recognize the same ligand (PS)
- Transduce opposing intracellular signals
| CD300a | CD300b | |
| Inflammation | ↓ | ↑ |
| Efferocytosis | ↓ | ↑ |
| ROS production | ↓ | ↑ |
| Neutrophil activation | ↓ | ↑ |
Efferocytosis (Dead-cell clearance)
- Recognition of the “eat me” signal (PS)
- Rapid clearance of apoptotic and dying cells
- Inhibition of sterile inflammation by limiting DAMPs release and subsequent PRR activation
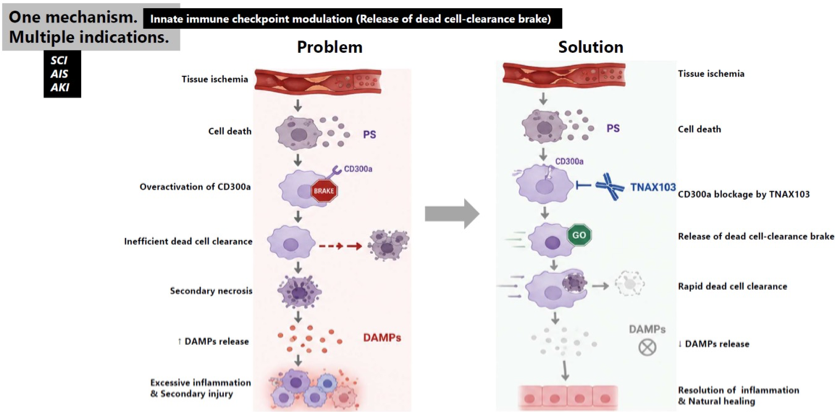
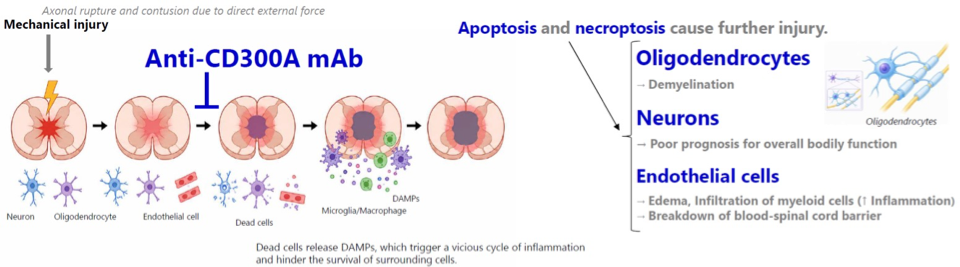
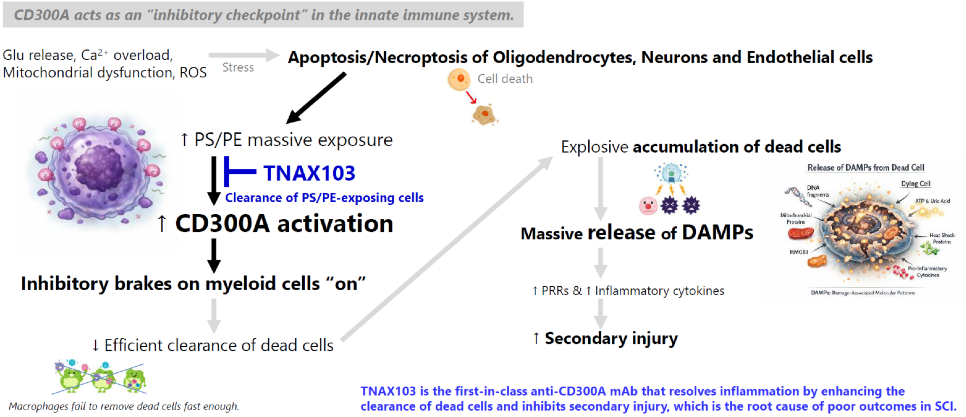
CD300a is an inhibitory receptor expressed on myeloid cells that contains an immunoreceptor tyrosine-based inhibitory motif (ITIM). Upon binding to PS expressed on dying cells, CD300a delivers inhibitory signals that act as a brake on innate immune cells, preventing excessive inflammatory responses. Although this inhibitory pathway protects healthy tissues from unnecessary inflammation, excessive CD300a signaling can also inhibit macrophage activation and efferocytosis (dead-cell clearance), allowing dead and dying cells to accumulate. When ischemic tissue is reperfused, these uncleared cells eventually undergo secondary necrosis and release DAMPs into the surrounding tissue and circulation. DAMPs are intracellular molecules that serve as endogenous danger signals. DAMPs are recognized by PRRs, which cannot inherently distinguish sterile tissue injury from microbial infection. As a result, innate immune cells rapidly become activated, producing an exaggerated inflammatory response characterized by neutrophil recruitment and tissue-damaging inflammation despite the absence of pathogens.
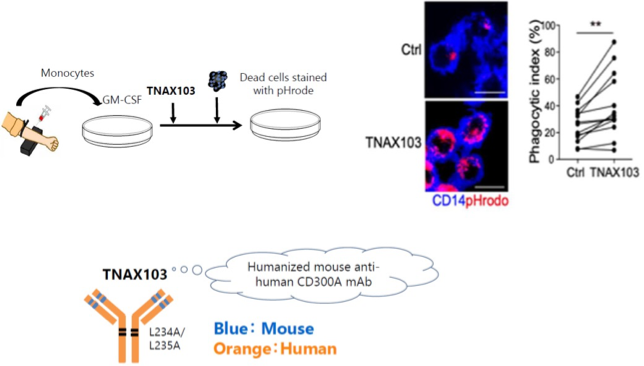
TNAX103 is a CD300A-neutralizing antibody designed to transiently release the inhibitory brake imposed by CD300A. By restoring efficient efferocytosis, TNAX103 enhances rapid clearance of dying cells “at an early stage”, limits DAMPs release, and helps the innate immune system respond appropriately to sterile tissue injury rather than amplifying unnecessary inflammation.
TNAX103 has the potential to become a “pipeline-in-a-pill” product expected to be applicable to multiple diseases through the same mechanism of action. TNAX103 enhances dead-cell clearance under ischemic conditions.
- Oligodendrocytes, Neurons & Endothelial cells → Spinal Cord Injury (SCI)
- Tubular endothelium → Acute Kidney Injury (AKI)
- Brain microvascular endothelial cells → Acute Ischemic Stroke (AIS)
SCI
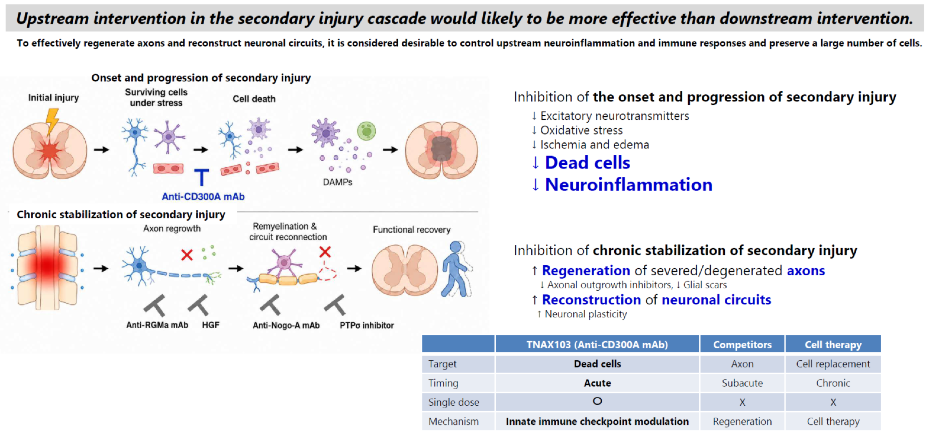
SCI remains one of the largest unmet needs in neurology because no approved therapy suppresses secondary injury.
Novel drugs inhibiting secondary injury, which is the root cause of SCI, are needed.
Anti-CD300A antibodies resolve inflammation by enhancing dead-cell clearance.
TNAX103 disables the innate immune checkpoint suppressing dead-cell clearance and resolves the vicious cycle of inflammation.
TNAX103 enhances efferocytosis (dead-cell clearance) by human monocyte-derived macrophages.
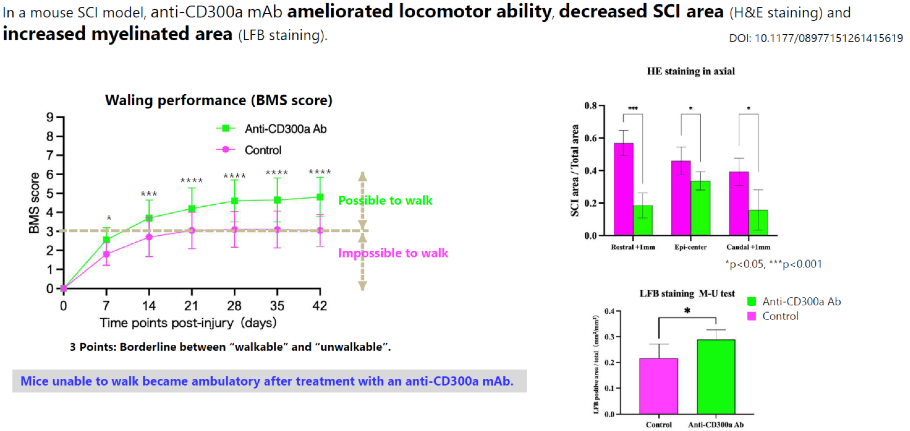
Anti-CD300a antibody ameliorated locomotor performance and histological findings in a mouse SCI model.
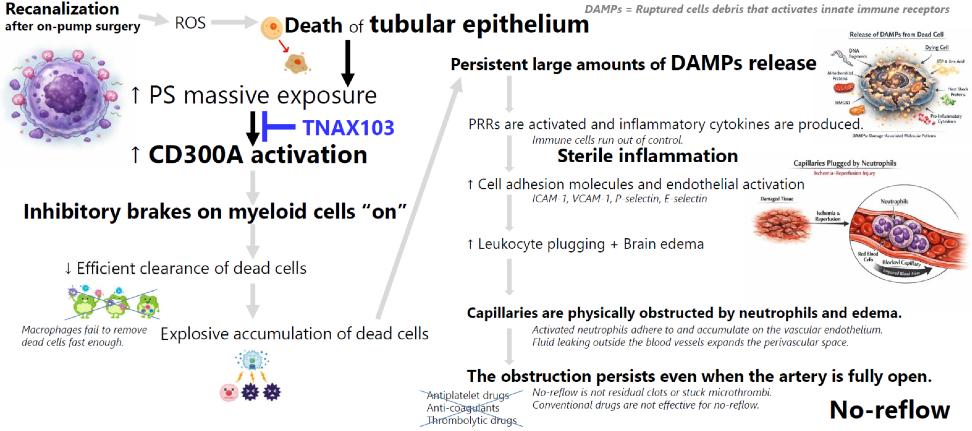
CSA-AKI
Over two million cardiac surgeries are performed annually worldwide, yet no approved drug prevents CSA-AKI.
TNAX103 reactivates myeloid cells and improves CSA-AKI by alleviating microcirculatory failure.
Anti-CD300a antibody enhances efferocytosis and decreases renal tissue injury, renal dysfunction and fibrosis.
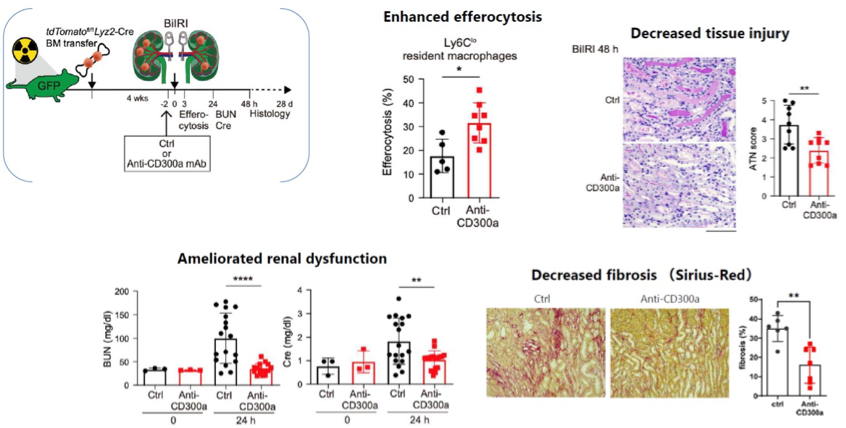
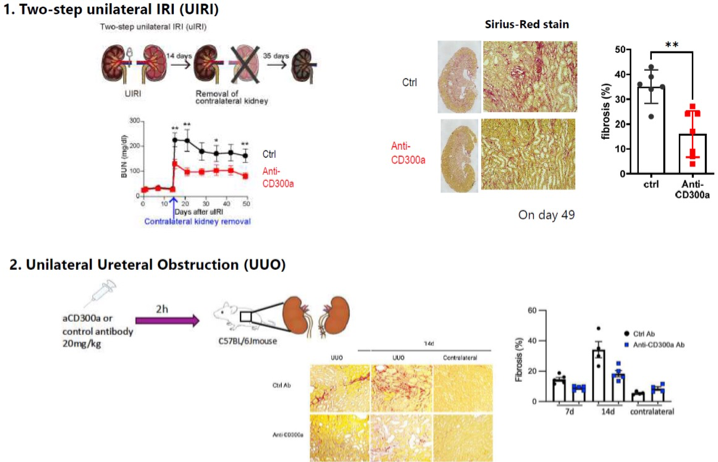
Anti-CD300a antibody ameliorates fibrosis in CKD models.
AIS
Despite successful reperfusion, 40-60% of patients remain disabled because of ischemia-reperfusion injury.

TNAX103 restores physiological tissue recovery while conventional therapies restore blood flow.

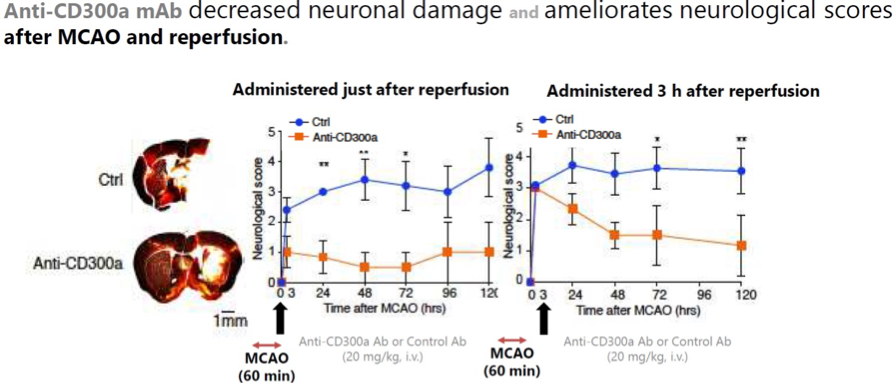
Anti-CD300a antibody ameliorates neuronal damage and neurological scores in an AIS and reperfusion model.